INTRODUCCIÓN
El Retinoblastoma (Rb) es la neoplasia maligna intraocular primaria más frecuente en la infancia. Supone entre un 1-3% de todos los tumores pediátricos. Es un tumor retiniano propio de la infancia .
Tiene una Incidencia (casos nuevos) entre un 1/15000-1/17000 de recién nacidos vivos. El 60% de los Retinoblastomas son esporádicos , el 40% hereditarios (15% unilaterales y 25% bilaterales).
La edad media al diagnóstico es más temprana en los bilaterales son los 12 meses y 23 meses en los unilaterales.
El Retinoblastoma tiene una tasas de supervivencia >90% a los 5años.
Existen distintos tipos de crecimiento; es un tumor que con frecuencia se calcifica (lo que también nos sirve de ayuda en el diagnóstico por imagen, como TAC, al ver el calcio). Puede diseminarse por contiguidad (extensión directa) a la órbita o cerebro, o por la sangre dando metástasis, sobre todo en médula ósea.
SIGNOS DE PRESENTACIÓN
• Leucocoria ( reflejo pupilar blanco): es la más frecuente
• Estrabismo (desviación del ojo hacia dentro o fuera)
• Inflamación ocular
• Glaucoma
• Celulitis orbitaria (edema intenso de párpados, con disminución de los movimientos oculares)
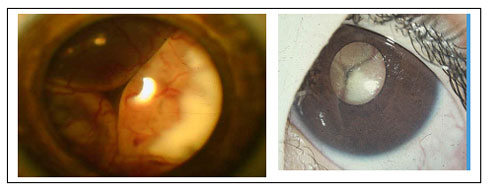
Imagen de un Retinoblastoma que ha producido un desprendimiento de retina, este niño a simple vista presentará un reflejo blanco de la pupila. Leucocoria
DIAGNÓSTICO
En primer lugar una buena exploración oftalmológica, atendiendo a los síntomas de consulta, a los antecedentes familiares, alergias…
Intentaremos según la edad del niño tomar la agudeza visual, exploraremos la motilidad de los ojos y bajo dilatación farmacológica con colirios veremos el fondo de ojo.
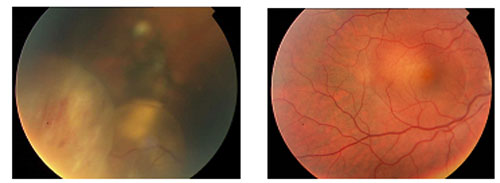
Imágenes de fondos de ojo compatibles con Retinoblastoma
Como pruebas complementarias solicitaremos:
- Ecografía.
- TAC.
- RM.
- Estudio de extensión a otros óranos, en estrecha colaboración con el Servicio de Oncología.
Los protocolos de actuación y baterías de pruebas complementarias varían según hospitales y según el estadío en que se encuentre el tumor (dependiendo de la localización retiniana, del tamaño tumoral, de si existe siembra vítrea o no, de la posible extensión extraocular, de si sólo hay un foco tumoral retiniano o varios y de muchos más factores que hay que considerar).
TRATAMIENTO
No existen unos criterios universales de tratamiento, es una decisión que se toma de forma conjunta entre los Servicios de Oftalmología y Oncología, siempre haciendo partícipe en la toma de decisiones a los padres del niño.
Señalaremos las diversas opciones de tratamiento sin entrar en las muy variadas indicaciones:
- Enucleación: Extirpar el globo ocular, junto a un trozo de nervio óptico. Se practica en casos unilaterales avanzados fundamentalmente. El contenido extraído en quirófano se envía al servicio de anatomía patológica, que nos informará si el fragmento de nervio óptico no está afectado, con lo cual el tratamiento se considera curativo; o si en el fragmento sí que encuentran células tumorales, con lo que será necesario un tratamiento adicional, bien sea radioterapia o quimioterapia. Posteriormente se colorará una prótesis. Es posible que al crecer el niño requiera un cambio de la misma.
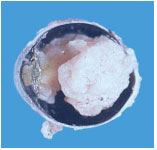
Pieza anatomo-patológica de un Retinoblastoma
- Radioterapia, sóla o como coadyuvante a otras terapias, actualmente se tiende a administrar en placas sobre el tumor para intentar su resolución y así preservar el ojo, aunque es necesario recordar que el retinoblastoma es un tumor que puede provocar el fallecimiento del niñó, por lo que siempre debemos pensar en el pronóstico vital.
- Quimioterapia, de entrada cuando el tumor es bilateral; también cuando el tumor es unilateral de gran tamaño y cumple los criterios establecidos, para reducir la masa tumoral y poder aplicar un tratamiento local, como la radioterapia en placas o braquiterapia, el láser u otros. De este tratamiento se encarga fundamentalmente el servicio de Oncología.
- Crioterapia: tratamiento local, se administra frío sobre la lesión tumoral, han de ser lesiones solitarias y generalmente periféricas.
- Termoterapia con láser diodo, es otra modalidad de tratamiento local, sobre lesiones solitarias, que pueden estar en polo posterior ( zona más próxima a la mácula o nervio óptico).
SEGUIMIENTO
Los protocolos de seguimiento varían igualmente según los centros. Los niños son seguidos de forma cercana, hasta la remisión de la tumoración, espaciando las visitas entonces.
Obviamente son seguidos por ambos servicios, y por aquellos que además fuese preciso. Los niños mas pequeños suelen ser explorados en quirófano bajo sedación, dada su escasa colaboración y la importancia de la revisión del fondo de ojo.
Las tasas de supervivencia son superiores al 90% a los cinco años. En los casos bilaterales el pronóstico empeora. En los casos unilaterales, el ojo no afecto es un ojo completamente normal. En caso de evisceración de algún ojo, los niños aprenden a hacer vida completamente normal con un solo ojo, desarrollándose en la vida con completa normalidad.
Dra. Emma Ausín González