La retinosis pigmentaria (RP) es un conjunto de enfermedades hereditarias de tipo degenerativo que afecta a la retina y se caracteriza por la reducción progresiva del campo visual, ceguera nocturna y pérdida progresiva de la agudeza visual. Es la 5ª enfermedad hereditaria más frecuente y la cuarta causa productora de ceguera.
La RP puede aparecer de forma esporádica o responder a un patrón hereditario, bien sea autonómico recesivo (requiriendo que ambos progenitores sean portadores), dominante o bien ligada al sexo. Por definición la RP es bilateral salvo en escasas excepciones.
En España el número de afectados supera os 15.000 individuos, estimándose en 480.000 los portadores del gen defectuoso y por lo tanto posibles transmisores de esta enfermedad a sus descendientes. Es ligeramente más frecuente en varones.
Es una enfermedad de curso progresivo que empieza en la niñez o en la adolescencia acentuándose después de los 20 años. Evoluciona de forma diferente según las personas y no conduce en todos los casos al mismo grado de pérdida de visión.
Afecta inicialmente a la visión nocturna ocasionando una lenta o deficiente adaptación a la oscuridad, existiendo dificultad para ver en lugares con poca iluminación. Es lo que se conoce como ceguera nocturna o hemeralopia. Comienza entonces el depósito de pigmento en la retina periférica (Figura 1), pero la persona afectada puede no ser consciente de estos cambios que reducen su campo visual.
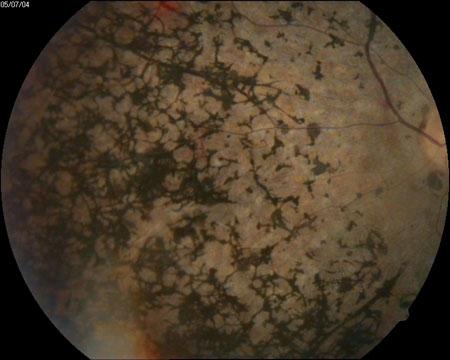
Figura 1
En este estado de la enfermedad la capacidad de vida y para el trabajo en general es buena aunque no pueden realizar actividades laborales nocturnas y/o en lugares con poca iluminación.
La enfermedad progresa posteriormente con una pérdida de campo visual mayor. Se agudiza la carencia de visión lateral que oscila aproximadamente entre los 30 y los 50 grados, hasta llegar a reducirse posteriormente al mínimo. La mayoría de las personas con RP grave quedan ciegas antes de los 40 años.
Los síntomas son la ceguera nocturna, el deslumbramiento (a los pacientes afectos de RP les molesta más la luz y es necesario el uso de gafas de sol especiales) , la reducción del campo visual y pérdida de agudeza visual.
El diagnóstico se establece cuando se presenta afectación bilateral, pérdida de visión periférica, mal funcionamiento y pérdida progresiva de los fotorreceptores. Las alteraciones del fondo del ojo (la retina) que consisten en la aparición de pigmento no tiene, aunque frecuentes, necesariamente que estar presentes para diagnosticar la RP.
El cambio esencial y primero de la retina es la degeneración y desaparición de los fotorreceptores (conos y bastones) , células encargadas de convertir el estímulo visual de la imagen en señales eléctricas que son enviadas al cerebro y allí son interpretadas. Los defectos genéticos identificados se localizan en estas células y en el epitelio pigmentario de la retina, capa de células que tiene un importante papel en la nutrición y mantenimiento de los fotorreceptores.
En el momento actual la RP no tiene tratamiento. Tan solo en aquellos pacientes con otras alteraciones asociadas con relativa frecuencia a la RP como son la presencia de catarata congénita o de edema macular cistoide (encharcamiento de la retina) podemos incidir discretamente en su visión, sin que modifiquemos de forma significativa el pronóstico visual a largo plazo de la enfermedad.
La posibilidad de llevar a cabo en un futuro más o menos próximo el trasplante de células del epitelio pigmentario de la retina o de los fotorreceptores ,tomados de retinas embrionarias o de cultivos celulares, abre una importante esperanza en el tratamiento de esta enfermedad.
Luis Amselem Gómez
Enlace a la asociacion de afectados de retinosis pigmentaria:
www.retinacv.com